



Raúl Pérez-Soto,a,d† Mihai V. Popescu,a† Sabari Kumar,a† Leticia Adao Gomes,b Changyeob Lee,a,c Elijah Shore, a Steven A. Lopez,b* Robert S. Paton,a* and Seonah Kima*
a Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA, b Department of Chem- istry and Chemical Biology, Northeastern University, Boston, Massachusetts, USA, c Department of Engineering, Korea Aerospace University, Hanggongdaehak-ro, Deogyang-gu, Goyang-si, Gyeonggi-do, Republic of Korea, d Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) CSIC- Universidad de Zaragoza, Zaragoza, Spain.
KEYWORDS(WordStyle“BG_Keywords”).Ifyouaresubmittingyourpapertoajournalthatrequireskeywords, provide significant keywords to aid the reader in literature retrieval.
Abstract:
The development of Graph Neural Networks for the task of predicting molecular properties has gained a great deal of attention as it typically allows the correlation of quick to compute atomic and bond descriptors with overall molecular properties. With the raising interest in photochemistry and photocatalysis as sustainable alternatives to thermal reactions, curation of virtual databases of computed photophysical properties for training of machine learning models has become popular. Unfortunately, current efforts fail to consider the exciton localization onto different chromophores of the same molecule, leading to potentially large prediction errors. Here we describe a molecular fragmentation strategy that can be used to overcome this limitation, while also providing a way to compare structural diversity between different libraries. Using a newly generated a database of 46,432 adiabatic S0-T1 energy gaps (ALFAST-DB), we compare its diversity against two datasets from the literature and demonstrate that a fragment-based delta learning approach im- proves model generalizability while achieving accuracies matching those of to traditional message passing graph neural network architectures (MPGNN).
Introduction:
With the re-emergence of photochemistry and photocatalysis as a major driving force of organic chemical synthesis at the start of the 21st century1–6, the development of highly accurate computational methods for the prediction of photophysical properties has been deemed as an essential driving force for the acceleration of the discovery of new chemical reactivity and development of new materials7–11. More recently however, development of machine learning based approaches have emerged as a promising alternative to highly computationally expensive quantum mechanical methods for the prediction of chemical properties, in turn used as an alternative to experimental data12–15. One reason for the rise of machine learning in the last two decades has been an increase in the amount of data available, along with easier access to the most novel model architectures. However, the reliability of any model is highly dependent on the quality of the data that was used to formulate the model, and thus, the very timeconsuming step of data curation and validation becomes the rate determining step in model development.
Due to the difficulties associated with collection of experimental data, in recent years models have relied on the construction of large datasets from computational simulations, based on methods such as Density Functional Theory (DFT)16. This approach sidesteps several of the limitations associated with data collection, quality assurance and data interpretation. A plethora of models trained on such computational datasets have been successfully developed with applications in many fields, including solubility17,18, bond dissociation energy19–21, synthesizability22, and catalytic efficiency prediction23,24. Thanks to the relative ease of constructing these virtual datasets, many research groups have constructed independent datasets for model development, frequently competing to simulate the same properties. This in turn raises a key question: how can we compare the diversity and quality of competing datasets beyond just the numerical range of the property in question? How can we improve new datasets based on the existing ones to cover broader swathes of the chemical space?
One such area with high interest in both the experimental and theoretical communities has involved the development and curation of large datasets of photophysical properties. Specifically, within the context of both photoredox and energy transfer catalysis, knowledge of the catalyst and substrate triplet energy is essential for the calculation of excited state redox potentials and also facilitates the prediction of reactions amenable to Dexter energy transfer. Within this regard, DFT computation of the
adiabatic triplet energy gap (i.e. the thermodynamic driving force between S0 to T1 state), has become a central method for determining molecular triplet energies, largely due to the experimental challenges involved. To this extent, the adiabatic triplet energy has become a key photophysical property, featured in numerous databases, such as Verde Materials DB25. Similarly, Glorius and coworkers have recently published the first machine learning model EnT-Decker for the prediction of adiabatic S0-T1 value, trained on an in-house generated DFT dataset of 34,848 molecules26. In this work, not only do they demonstrate that Graph Neural Network models outperform other simpler models but also showcase the practical application of the developed models and build a user-friendly platform for chemists to use their models.
However, as it will be demonstrated in this work, highthroughput DFT calculations of adiabatic S0-T1 gaps presents challenges when applied to molecules with multiple functional groups. In such cases, exciton localization can vary semi-randomly across different regions of the molcule, leading to significant energy differences and introducing substantial error in model construction. In this work, we present a new fragmentation algorithm designed to address this issue. This algorithm enables the training of a ∆-learning model on a new dataset of 46,415 molecules (ALFAST-DB), following a similar protocol to EnT- Decker. Additionally, the algorithm provides a novel way to compare the chemical composition of different data-bases and evaluate the transferability of ML models trained on them.
Exciton Localization Problem in Multichromophore Systems:

Figure 1: Diagram describing the computational structural optimization on the T1 surface starting from a ground state geometry
As highlighted earlier, DFT optimizations on excited states are particularly challenging for molecules with multiple chromophores due to the issue of exciton localization. Taking allylbenzene as an example, the standard approach begins by using the optimized geometry of the molecule in its ground state (S0) as the starting point for optimizing the corresponding triplet state (T1). The geometry optimization on the T1 surface proceeds from this initial geometry, and the algorithm directs the structure toward the nearest local minimum based on the gradient of the surface curvature.
In the case of allylbenzene, this initial optimization leads to a structure where the triplet diradical localizes on the phenyl group, as illustrated in Figure1. However, this localization isn’t always unique. If the molecule is initially excited to a higher triplet state (Tn) instead, the system may follow a different potential energy surface. This new surface can intersect the initial T1 surface and, in some cases, drop below it in energy, effectively becoming the new T1 state. Such a scenario results in the localization of the spin density on a different region of the molecule.
For allylbenzene, the system can alternatively find a T1 minimum where the spin density localizes on the alkene group. While the optimization process preferentially leads to the excitation of the aryl ring, this alkene-localized minimum is also a valid solution. In fact, it is energetically more favorable, resulting in a lower adiabatic triplet energy. In this case, when computed at the at the M06- 2X/def2-TZVP level using Gaussian 1627, the adiabatic triplet energy (62.4 kcal/mol) is lower by an impressive 23.3 kcal/mol compared to the phenyl-localized minimum (85.7 kcal/mol).
This difference in localization leads to a significant challenge when it comes to automated excited state optimization for database construction. In systems with multiple chromophores, it becomes nearly impossible to predict a priori where the triplet state will localize. As a result, this uncertainty complicates the construction of a coherent dataset, often leaving the data effectively “unlabeled” in terms of which chromophore is involved in the triplet state localization. This kind of unstructured data introduces confusion into machine learning models, as the models may struggle to learn from or differentiate between cases where different chromophores are involved. Furthermore, it raises an important question regarding the goal of adiabatic triplet energy predictions: is the objective to predict the global minimum energy state, or should the models aim to predict all possible local minima? Defining this objective becomes crucial in ensuring the predictive power and applicability of the machine learning models.
One potential solution, as implemented by the EnT- Decker authors, is to provide a prediction of spin density alongside the adiabatic triplet energy value. This strategy adds valuable information to the user, offering insight into where the triplet localization occurs within the molecule. However, while this represents an important step forward, it does not fully resolve the underlying issue of training models on unlabeled data. Since the spin density localization is not systematically assigned to a specific chromophore or fragment, the model is still learning from data that may not be explicitly categorized.
Moreover, since these spin density models were trained independently of the energy predictions, there is the po- tential for inconsistencies. In some cases, the energy and spin density predictions may not align, potentially leading to misleading conclusions for the user.
Molecular Fragmentation Algorithm
To address the challenges associated with exciton localization and un-labelled data in multi-chromophore systems, a first step is to develop a workflow for the identification of potentially relevant sites where the exciton can be localized. Here we developed a fragmentation algorithm that generates chemically meaningful fragments from complex molecules, akin the one described by Ertl in 201728. This approach was inspired by the conxcept of functional groups in organic chemistry, which serve as the building blocks of molecules and represent key sites of chemical reactivity. Functional groups, often composed of π-bonds and heteroatoms linked by resonance, are rich in π-electrons and lone pairs. These electron-rich regions are frequently the most likely sites for excitations due to their lower energy gaps between π-π* and n-π* orbital levels29. By leveraging this understanding of functional groups, this algorithm systematically breaks down molecules into these reactive components based on extended conjugation paths, leading to more structured data that can be effectively used for machine learning. The steps of the algorithm are illustrated below (Figure 2), and include the following key processes:

Figure 2: Scheme and example of the application of the fragmen- tation algorithm used in the present work.
- Identify all relevant heteroatoms based on a preefined list as well as all C containing a double, triple or aromatic bond. Specifically for this work we considered as candidate heteroatoms N, O, S, P, Se, F, Cl, Br.
- For each one of the candidate atoms, a list is contructed that contains these atoms. Then, the atoms they are directly bonded to are recursively added to each candidate atom. If the connecting atoms are also candidate atoms, the process continues to add their connected neighbors until no can- didate atoms are found. This recursive process is stopped when either there are no more atoms or the last atom added was a non-candidate atom.
- Once no candidate atoms remain in reach molecule, duplicated fragments are removed.
- Finally, for database storage analysis and curation, each fragment is transformed into its SMILES representation. During this step the end-atoms of the fragments are implicitly capped with H to match their valence. As all valid SMILES are valid SMARTS, H-capping allows the generation of smaller, valid, typically closed-shell molecules containing the key functionality of the fragment.
Following the example highlighted above (Figure2), the outcome of the fragmentation scheme as applied to 3- (2-(4,5-dichlorocyclohex-2-en-1-yl)ethyl)-1,1-dime- thylurea will lead to the identification of four distinct func- tional groups, namely: a trisubstituted urea, a disubstituted (Z)-alkene and two alkyl chlorides.
Databases Overview and Curation
The present work makes use of three separate datasets comprising of adiabatic S0-T1 energy gaps. One such dataset is the EnT-DB database developed by Glorius and coworkers used to develop the EnT-Decker predictive model. The EnT-DB dataset consists of DFT-computed adiabatic S0-T1 energy gaps for a total of 34,848 molecular species. Energies are computed at the ωB97x-D3/def2- TZVPP;PCM(CH3CN)//B3LYP-D3/def2-SVP;
PCM(CH3CN) 30–35 level of theory, using the ORCA software package36. Molecules were selected to maximize chemical space coverage of medicinally relevant structures, and selected from the ZINC dataset 37clade of commercially available species with molecular weights of under 325 Da. The size distribution of molecules in the dataset is roughly symmetric, with a median of 30 atoms. Calculated adiabatic S0-T1 gaps span a broad range, with a median of 69.4 kcal/mol (Figure 3, EnT-DB).
The second database used in this work is the Verde Materials Database previously developed by Lopez and coworkers (Verde-DB). The version of the dataset used here is more comprehensive than that published previously; this version of the Verde-DB dataset contains 3286 molecules, as compared to the 1,500 molecules in the original work. The S0-T1 adiabatic gaps present in this daabase are also obtained using DFT, but computed at the M06/6-31+g(d,p), PCM(CH3CN)38–42 level of theory. In contrast to EnT-DB, which only focuses on adiabatic S0- T1 gaps as a target property and prioritizes a broad chemical scope, Verde-DB is more focused on broader coverage of properties related to photoredox catalysis, such as excited state reduction and oxidation potentials or S0-S1 energy gaps, at the cost of a narrower chemical scope. This chemical scope is more focused on molecules used in commercial applications, with additions of computationally generated derivatives. The molecular size distribution (Figure 3) has the same median as the EnT-DB dataset (30 atoms), while showing a pronounced tail towards larger molecules. Due to the specialized chemical scope targeted in the Verde-DB dataset, it exhibits a strong bias towards molecules with a small S0-T1 adiabatic gap, with a median value of 28.2 kcal/mol.
Finally, we develop our own database targeting a broad chemical scope, following a protocol where optimization of the T1 geometry is carried out using the optimized
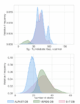
Figure 3: Total number of atom and S0-T1 adiabatic gap distributions of the 3 presented datasets.
ground state structures as a starting point. Previously, we compiled a large database of DFT-computed structures for the prediction of bond dissociation enthalpies, leading to the creation of the ALFABET model. The ALFABET dataset was designed to cover a broad range of organic chemical space, and comprises molecules with C, H, N, O, S, Cl, F, P, Br, and I atoms. Building on this previous work, we compute the S0-T1 adiabatic gaps for molecules of interest in this database. This initial set comprises 57,736 molecules after removing radicals and retaining only those molecules with double, triple, or aromatic bonds. For the level of theory used in computation, we utilize the M06-2X/def2-TZVP level of theory, in contrast to that used to construct the EnT-DB dataset: although the M06-2X functional had been identified as optimal for adiabatic triplet energy calculations by the Glorius group, challenges with convergence and computational costs led to the ωB97x-D3 functional being used instead. While recent work suggests that benchmarking of adiabatic triplet energies against experimental measurements may not be representative of physical reality and can lead to very large errors on systems that undergo high structural reorganizations, benchmarking against the adiabatic values computed at the “gold standard” DLPNO-CCSD(T) method with the cc-PVTZ basis set 43–48also showed that the M06-2X provides the best accuracy. Further, the S0 ground states in the original ALFABET dataset were computed at the M06-2X/def2-TZVP, allowing us to avoid incurring the large computational cost mentioned by Glorius and coworkers; we thus computed the T1 minima at the same level of theory. Out of the 57,736 starting molecules, 53,137 achieved successful convergence, which were
further curated down to 46,415 molecules after further quality assurance checks (see SI for further details). As seen in Figure 3 this database is more focused towards smaller molecules than the EnT-DB or the Verde-DB datasets, with a median number of atoms of 18. However, we maintain a similar distribution of S0-T1 adiabatic gaps to EnT-DB, with a median of 72.6 kcal/mol. We refer to this new dataset as the ALFAST-DB.
Database Analysis and Comparison
Using the framework of the proposed fragmentation scheme, we analyze the fragment compositions of the three databases discussed above. First, fragments that did not contain a double, triple or aromatic bond (using SMARTS pattern matching) were discarded. These frag- ments were then used to obtain the possible exciton lo- calizations within each molecule across the databases (vide infra). This fragment-based breakdown allows for a more quantitative evaluation of the relevant chemical di- versity and overlap between the three datasets.
| Databases | ALFAST | EnT | Verde |
| ALFAST | 15,410 | 2,146 | 0 |
| EnT | 2,146 | 18,909 | 0 |
| Verde | 0 | 0 | 3,286 |
First, we evaluate the fragment overlap between data- bases (Table 1). We observe that while the ALFAST-DB has a larger number of molecules (46,415 vs 34,848), it has a slightly smaller diversity of unique fragments than the EnT database (15,410 vs 18,909). Between these two databases there are 2,146 shared fragments, while neiher database contains shared fragments with the Verde- DB. Next, we analyze the number of fragments formed for each molecule and quantify the percentage of shared fragments within this classification scheme (Table 2). Interestingly, a large number of molecules in each dataset contain a fragment from the small shared subset of 2,146 fragments – 62.6% and 51.6% for ALFAST-DB and EnT- DB respectively, indicating a chemical space overlap. This overlap is thus more significant than one would expect if only the identity of the complete shared molecules (of which there are 1530) are considered. Almost all unique fragments correspond to single-fragment molecules, and the EnT-DB has a higher composition of mult fragment molecules (32.4% vs 15.5%). Notably, the Verde-DB is constituted solely of mono-fragment molecules (thus not shown for brevity). We posit that this may be a consequence of the more specialized scope considered when constructing the Verde-DB.
It is important to note that these statistics do not provide a basis for claiming the superiority of one dataset over the other. On one hand, a larger number of unique fragments can be taken naively to represent increased chemical diversity of the constituent chromophores; while on the other hand, a larger ratio of molecules to unique fragments provides a larger diversity of substituent effects on the chromophores, thus potentially improving the generalization characteristics of derived models.
Table 2: Molecule breakdown of the ALFAST-DB and EnT-DB. Molecules are classified as either single-fragment or multiple- fragment, and based on if at least one of the fragments is within the subset of fragments shared across the two databases (Shared) or all fragments of the molecule are unique. Numbers in parenthesis are the absolute number of molecules.
| ALFAST-DB | EnT-DB | |||
| Single | Multiple | Single | Multiple | |
| Unique | 37.2% (17,289) | < 0.1% (35) | 47.8% (16,649) | 0.6% (212) |
| Shared (2,146) | 47.2% (21,941) | 15.4% (7,150) | 19.8% (6,911) | 31.8% (11,076) |
| Total | 84.5% (39,230) | 15.5% (7,185) | 67.6% (23,560) | 32.4% (11,288) |
Finally, with the aim to assess the effect of exciton localization mentioned previously, we utilize the Mulliken spin densities at the optimized T1 states included in the ALFAST-DB to compute the total spin of each fragment obtained. The fragment with the highest spin was then

Figure 4: (A) Absolute (blue histogram) and cumulative frequency (black line) of the distribution of the main fragment’s spin within the ALFAST-DB. (B-D) Example DFT-computed S0-T1 adiabatic gaps for fragments and for multi-fragment molecules within the ALFAST-DB and the EnT-DB. The spin of each fragment was computed from the atomic Mulliken Spin Densities. The example fragments (B) were computed at the same theory level as ALFAST-DB.
selected as the “main fragment” of the molecule, effectively labeling each molecule with a main fragment, and a total spin of the main fragment. The distribution of these main fragment spins across the ALFAST-DB is shown in Figure 4A. While the majority of the database has a clearly localized triplet, defined as where the spin of the main fragment is over 1.7, there are three notable outlier regions that require further discussion: molecules that have main fragment with a spin over 2, molecule where the main fragment spin is between 0.7 and 1.3, and molecules whose main spin is below 0.65. We posit that the first region emerges as an artifact of the truncation during the total spin density calculation of the fragment. The second and third regions, while having more consequential root causes, constitute less than 1% of the molecules of ALFAST-DB (less than 500 molecules), and thus their im-act on the overall database is taken to be minor. The second region (between 0.7 and 1.3) corresponds to molecules where a charge transfer state was likely obtained. The third region, however, emerges as molecules whose main fragment was incorrectly assigned. This emerges in cases where the obtained minima in the T1 surface was not localized in a fragment containing a double, triple or aromatic bond.
To illustrate this electron delocalization problem further, we analyzed representative examples from each outlier region described above (Figure 4B, C) Examples were chosen such that the localization of the exciton can be confirmed based on the spin of the fragment. In addition, some example molecules where a charge transfer state was obtained are highlighted (Figure4D). It can be seen in Figure 4B that the adiabatic S0-T1 gap of toluene is lower than the adiabatic gap of acetonitrile. As such, a molecule containing both moieties separated by an alkyl carbon chain should be able to access the triplet state of both moieties. In the selected examples (ALFAST-DB and Ent-DB, left column, Figure 4C) the computed value is more similar to the adiabatic gap of toluene, and in the ALFAST-DB example we can confirm that the exciton location is the phenyl ring. However, in this example, the overall computational protocol can be considered successful on the task of approximating the adiabatic S0-T1 gap, understood as the lowest free energy difference tween the S0 free energy surface and the T1 free energy surface.
On the other hand, the examples highlighted with phe-nyl and alkene moieties (ALFAST-DB and Ent-DB, right column, Figure 4C) show the opposite result: out of the two exciton locations the minima with the phenyl-localized triplet was favored, which is higher in energy than that localized in the alkene (based on the difference between toluene and propene fragments), thus illustrating an unsuccessful application of the fragmentation protocol.
Further work is necessary to thoroughly analyze this discrepancy; however, due to the relatively small number of such examples, we simply remove them from the database here.
The final charge transfer examples (Figure 4D) show two different cases. In the rightmost one, two clear disconnected π-systems where the spin is localized, while the other example shows a case where not all the spin is localized in a region of the molecule containing a double, triple or aromatic bond. As before, such fragments are omitted from consideration.
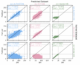
Figure 5: Model extrapolation across datasets. Parity plots of adiabatic S0-T1 predictions in kcal/mol of each dataset (column) of the same model architecture trained each dataset (row). For example, parity plot “ab” corresponds to the architecture trained with the combined training and validation set of l ALFAST-DB (80% of the database) and shows the prediction of EnT-DB.The predictions on the diagonal plots only shows the molecules in the test set (remaining 20% of the database) of each respective database. The metrics, R2 and MAE values, correspond to the data presented in each plot. MAE values are in kcal/mol.
Architecture Selection and Model Extrapolation
Prior work by numerous groups has shown the effectiveness of Graph Neural Network models in predicting molecular properties. Here, we tested different message passing neural network architectures based on the previously successful ALFABET model (further details can be found in the supporting information). Model evaluation was performed using a 64:16:20 train:validation: test split of the ALFAST-DB with randomized splitting. A 5-fold cross-validation strategy was used within the training-validationtion subsets. The validation loss function (mean absolute error, MAE) was tracked during training and used to select the best per-validation fold. The final architecture and training strategy presented a cross-validation MAE (aggregated across folds) and test-set MAE (averaged across folds) of 1.94 and 2.00 kcal/mol, respectively. For the final predictions and the presented parity plots, the
model was trained using the combined training and validation sets, resulting in a test-set MAE of 1.94. When trained on the EnT-DB using a similar splitting strategy, the cross-validation MAE and averaged test-set MAE were 2.04 and 2.06 kcal/mol, and a final model test-set MAE of 2.37. Compared to the random splitting results of the EnT-Decker GNN models, with an MAE of 1.97 and 1.93 kcal/mol for the Chemprop-D-MPNN and AttentiveFP-GNN models, respectively. It can be observed that our selected architecture achieves similar accuracies, independent of the database it was trained on.
After the architecture selection, the performance of the models on the other unseen datasets was evaluated to assess model extrapolation capabilities. As each database is generated using a different DFT level of theory, a degradation in model performance is to be expected. This increase in model error stems from two separate causes: the uncertainty of the ML model in making predictions on unseen data, and the error incurred when comparing S0-T1 adiabatic values computed using different DFT theory levels. To attempt to separate these two sources of error, each database’s final model (trained with the respective combined training and validation sets) was then used to predict the values of each dataset (Figure 5). The prediction of each final model on the test set of its respective database (Figure 5 aa, bb, and cc) provides an estimate of the model uncertainty. Conversely, the predictions on the complete unseen datasets (Figure 5ab, ac, ba, ca, bc and cb) provide insight into how the model extrapolates.
From this analysis, two clear trends are observed. First, when the architecture is trained on either ALFAST-DB or EnT-DB (ie., the datasets with more general chemical space coverage) and used to predict the other, (Figure5 aband ba), a clear increase in the MAE is observed while some degree of correlation (marked by the R2) is retained. On the other hand, when the training and prediction are performed using the more specialized Verde-DB and the other two remaining databases (Figure 5ac, bc, ca, and cb), respectively, the models fail drastically at extrapolation. This behavior reproduces known trends regarding the importance of training set chemical space diversity on model performance.
To estimate the magnitude of the error incurred when comparing values computed with different DFT functionals, we analyze shared molecules between ALFAST-DB (in gas phase) and EnT-DB (in acetonitrile). For this task, the canonical SMILES representation of molecules from both databases was obtained and used search for common molecules, of which 1,530 were found. An MAE and R2 of 3.36 kcal/mol and 0.88 respectively were obtained when comparing the S0-T1 adiabatic gap of these 1,530 molecules (see Supporting Information); a linear fit was also performed, yielding an MAE and R2 of which stayed as 2.63 kcal/mol and 0.92 respectively (see Supporting Information). It should be noted that this MAE represents errors derived from directly comparing the results of different DFT methodologies, but also molecules where each method computed a different exciton location, molecules whose exciton is located on the same fragment but geometry optimizations led to different local minima, or molecules that would be more drastically stabilized in the triplet state by the acetonitrile solvent.
If then, the uncertainties in both model predictions and the underlying DFT methodology are combined, the model trained on ALFAST-DB achieves a net uncertainty of 5.30 kcal/mol, which is comparable to the MAE of 5.51 kcal/mol obtained when using the same model to make predictions on the EnT-DB (Figure 5 ab). The same can be observed when the model trained on the EnT-DB extrapolates to the ALFAST-DB. This observation stays the same even when the cross-validated MAEs of the models are considered (1.94 and 2.04 kcals/mol for ALFAST-DB and EnT-DB respectively).
Contrary to this, we see that such “symmetric” behavior is not observed when considering the Verde-DB. As seen from the analysis above, the Verde-DB shares no fragments nor molecules with either of the other two

Figure 6: Boxplots with total (all) and marginal absolute error distributions of molecules based on whether they have a shared fragment between databases (shared vs non-shared) or whether they are mono- or multi-fragmented molecules (mono vs multi). Q1 and Q3 are the first and third quartiles, respectively, and IQR stands for interquartile range. The error distributions of the dataset used for training are the distributions of the database’s test set, while the distributions for the extrapolated database include the full extrapolated database.
databases, is constituted only of mono-fragment molecules and is an order of magnitude smaller than the other two databases due to its more specialized nature.
Both the lack of shared fragments and the number of mono-fragment molecules may be responsible for the poor extrapolation to and from Verde-DB; thus, to provide further insight on this, a detailed breakdown of errors in the ALFAST-DB-trained and EnT-DB-trained models (Figure 6) based on fragment presence was obtained.
First, it is reaffirmed that the absolute errors are much smaller and the distribution narrower (Figure 6)when predicting the dataset that the model was trained on, as compared to the database it is being extrapolated to. However, a clear difference between the shared and non-shared fragment error distributions of the extrapolated dataset (Figure 6)can be seen, where molecules that do not have a shared fragment have a larger error than the molecules that have a shared fragment. This involves a shift of the median of approximately 1-2 kcal/mol and a broadening of the error distribution. On the other hand, such consistent effects are not observed between the mono vs multi-fragment distributions; interestingly, both models have a slightly better performance for multi-fragment molecules than for mono-fragment molecules, independently of the predicted dataset.
One interpretation of this trend is that the model better fits multi-fragment molecules as they are more information-dense. If indeed this is the case, the model should also exhibit lower errors for molecules that suffer from the exciton localization problem discussed earlier. It is to be noted that this interpretation assumes that the differences between the mono-fragmented and the multi-fragmented error distributions are not an artifact of the subset size (see molecule breakdown in Table 2), nor due to the lower chemical diversity of the main fragments in the multi-fragment molecules. Nonetheless, the consistent difference when extrapolating trained models to molecules with shared fragments vs molecules with non-shared fragments, and the poor extrapolation to and from Verde-DB point towards the shared-fragment overlap as the predominant cause of the large prediction errors.
It is thus clear that one method to improve the generalizability of a model trained on a dataset of properties dependent on the exciton location to increase the chemical scope of the fragments, instead of increasing the frequency of the fragments already present in the dataset.
Fragment-Based Models
As fragment overlap between different datasets seems to directly correlate with the model’s extrapolation ability, at a first-level approximation, it may be assumed that expansion of the datasets with more fragments or even using the fragments as a surrogate prediction for the entire molecule may provide an interesting approach towards improving the model performance.
To explore this angle, we created a dataset dubbed Fragments-DB, which consists of DFT-computed adiabatic S0-T1 gaps for each fragment present in ALFAST-DB or EnT-DB. We applied the fragmentation algorithm and included entries that are present at least twice within either of the two datasets, leading to a total of 6,058 unique fragments. Upon following the same protocol for the construction of ALFAST-DB, we obtained a total of 5135 S0-T1 adiabatic gaps for structures in Fragments-DB.
Using this new Fragments-DB, we retrained the GNN model following the same approach as employed prior (Figure5) in order to allow for direct comparison with the larger ALFAST dataset. Following this approach, the resulting GNN model was found to perform slightly worse, leading to a test-set MAE of only 2.98 kcal/mol and a coefficient of determination of 0.83 (Figure 7A).
The decrease of predictive accuracy when compared with the ALFAST-DB trained GNN (Figure 5aa, 1.94 kcal/mol) is attributable to the composition of Fragments- DB. It is to note that due to the construction of Fragments- DB, only one instance of each fragment is present in the
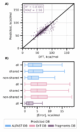
Figure 7: A) Predicted vs Computed S0-T1 adiabatic gaps of the model trained on the Fragments-DB. B) Boxplots with total (all) and marginal absolute error distributions of mole- cules based on whether they have a shared fragment in Frag- ments-DB (shared vs non-shared)
training set. Thus, a random splitting strategy does not have the same effect on model training with Fragments-DB- DB as with ALFAST-DB. The accuracy presented here is should hence be evaluated against the 3.12 kcal/mol MAE of the out-of-sample splitting results presented by Glorius26.
Intriguingly however, when the Fragments-DB-trained model was applied to predict the test set of ALFAST-DB, the R2 and MAE were found to be 0.72 and 5.36 kcal/mol, worse than the Fragments’ test-set MAE (2.98) or its cross-validation values (MAE of 3.07 and 3.26 for the aggregated validation and test set respectively, see SI). We then attempted to investigate the cause of this discrepancy by analyzing the errors in the context of fragment overlap (Figure7B). Notably, the errors remain consistent between shared and non-shared fragments, for both ALFAST-DB and EnT-DB. There are a number of possible explanations for this behavior: a) the degradation in performance may be due to the excitation localization problem discussed previously, as the fragment-based

Figure 8: General illustration of the different “prediction” schemes discussed (A-E left) and their corresponding parity plots on the test set of ALFAST-DB (A-E right). All presented numbers are in kcal/mol except the R2 coefficients which are dimensionless. In this figure “Core” is used to refer to the fragment where the exciton is localized. Reported MAE and R2 are the metrics of the predictions of the ALFAST-DB test set.
model is trained on individual chromophores only, and thus multi-chromophore molecules are out-of-distribution. .b) the model may be failing to learn contributions from the whole molecule (videinfra, Figure8D); c) the model fails at representing large molecules; d) it is known that certain functional groups more readily optimize to triplets than others (e.g. aryl groups); thus, it may be that the wholemolecule ALFAST model simply learns the identity of these groups more readily than the fragment model. We leave further investigation of these possibilities to future work.
In order to solve the exciton localization problem while also trying to explore the origin of the deviations between these models, we decided to utilize a series of new training strategies systematically considering the contributions of the fragments. We initially investigated the simple strategy of dataset augmentation, where ALFAST-DB was naively augmented using Fragments-DB (Figure), keeping only the entry from the Fragments-DB when a molecule was present in both databases. When trained on the combined training and validation sets of each database, a test-set MAE (evaluated only on the test-set of ALFAST- DB) of 2.18 kcal/mol and an R2 of 0.93 was obtained (cross-validation results and performance on the combined test-set are provided in the SI). When compared with the model trained on solely whole molecules from the ALFAST-DB (MAE of 1.94, Figure5aa), a small decrease in performance is observed. The resulting 11% degradation is attributable to the differing chemical spaces represented by ALFAST-DB and Fragments-DB: the Fragments-DB is composed of smaller constituent chemical species and thus presents a different input space distribution than the ALFAST-DB. Nevertheless, this new model forms a more robust baseline for performance comparison.
We next evaluated the incurred error when the adiabatic S0-T1 gap of the molecule is approximated as the adiabatic S0-T1 gap of the main fragment. As such, we developed a “core-fragment DB lookup scheme”, where we approximate the value of the S0-T1 gap for the whole molecule to be same as the DFT-computed value of the minimum size representation of the fragment where the excited triplet is localized (Figure 8C), providing an estimate of the error only due to the fragmentation approximation. Subsequently, the “core-fragment prediction scheme” utilizes a similar methodology; however, the Fragment-DB trained GNN model is used for the prediction instead of DFT (Figure 8D). Interestingly, for the GNN-based approach, the ALFAST-DB test set MAE is reduced from 5.36 to 3.01 kcal/mol by merely switching the target of the prediction from the full molecule (Figure 8A) to the main fragment of the molecule (Figure 8D). The MAE further decreases to 2.30 kcal/mol when the DFT-calculated fragment adiabatic S0-T1 gaps are used (Figure 8C). This observation is suggestive that, to a first-order approximation, the calculation and prediction of just the fragment bearing the “correct” excitation can provide a reasonably accurate prediction.
Upon further analysis of the outliers of these approaches, it is found that the remaining part of the molecule can in some instances have very large effects. In particular, it was found that most such outliers consist of fragments that were originally part of ring systems, which is consistent with previous studies (for example, a (Z)-butene formed from the fragmentation of cyclopentene, for which errors can be as high as 9 kcal/mol)7. While this observation is suggestive that the final predictions could be improved by further refining the fragmentation scheme, an alternative and more robust approach would be development of a model architecture that is able to learn the specific contributions of the neighboring environment not encapsulated by the fragment itself.
In light of these results, we attempted to use a ∆-learning scheme to further reduce model error. Here we keep the same core-fragment prediction scheme detailed above but adapt the architecture and the model training (detailed in the Supplementary Information) to take as inputs both the molecule and the core and output two values: one associated to the adiabatic S0-T1 gap for the provided core and a second value to account for the conribution of the rest of the molecule. Historically, ∆-learning models have been used to learn error corrections, aallowing low-level of theory quantum mechanical methods to be matched with results from more accurate computational or experimental methodologies49. This methodology has proven to be successful at predicting diverse chemical properties, such as solvated ground state redox poentials50, protein-ligand binding affinities51, and dielectric constants52. Here, we treat our core-fragment prediction model as a “base prediction” and predict a correction to it from the non-core atoms and bonds, thus considering atom contributions from the non-fragment constituent atoms. This adapted model follows a two-stage training (fully detailed in the Supplementary Information) where the Fragments-DB is used during the first training stage, to train the core-fragment prediction model weights and then a second stage where the combined ALFAST and Fragments-DB are used to fine-tune the final predictions.
Pleasingly, upon implementation and training of this ∆- learning approach, a test set R2 and MAE of 0.93 and 2.24 kcal/mol was obtained on the ALFAST-DB test set (Figure8E), without a large degradation in performance over the simplistic augmentation scheme (R2 = 0.93, MAE = 2.18 kcal/mol; Figure8B). More intriguingly however, the results obtained for the last approach are comparable in accuracy with the performance of the GNN trained on and applied to predict exclusively single fragment molecules (Figure5ccand Figure7). As such, we can conclude that this approach not only removes the excitation localization problem, but also is able to partially account for electronic effects contributions of non-fragment constituent atoms. While further investigations are needed to further elucidate the precise quantitative role of the non-fragment constituents, this approach leads to a significant improvement in performance when compared to the Ent-Decker and our original training approaches, providing a new baseline against which future GNN architectures for the predication of excited state properties should be evaluated.
Conclusions
In this work, a fundamental problem in the context of photoredox and energy transfer catalysis is identified: the localization of the exciton. With this problem in mind, we build accurate high-throughput computational databases for the to develop a machine learning models to predict
the adiabatic S0-T1 energy gap for small molecules. To do this, a chemically guided fragmentation algorithm was developed to address database analysis and curation. Next, its application on previously existing databases was showcased, highlighting the impact of exciton delocalizationtion on both previously developed and newly built databases. By analyzing the impact of the fragmentation scheme on the EnT-DB, ALFAST-DB, and Verde-DB datasets, and the subsequent changes in performance of GNN models trained on these datasets, principles for the design and augmentation criteria for fragment-based datasets targeting the adiabatic S0-T1 gap was elucidated. To improve database quality, the addition of diverse excitation localization moieties, “main fragments”, is preferable to the addition of duplicated versions of the same moieties, even when such moieties originate in diverse molecular species. Finally, based on these results, a promising strategy for the development of ML models to predict the adiabatic S0-T1 gap was developed, where the prediction of the adiabatic S0-T1 gap of the key chemical moiety for the target molecule is decoupled from the rest of the molecule. For this strategy to be valid, the property of the molecule must be well approximated by the property of the minimal motif/fragment. We show here that such a model has a similar error (MAE of 2.3 kcal/mol) to complex machine learning models trained on much larger databases (in the range of 1.93 2.26 kcal/mol). We believe that the present work marks a clear direction for the development of newer fragment-based models and databases in the field while also highlighting the importance of including domain knowledge in the prediction pipeline and the data analysis. Although the final approach with a fragment-based ∆learning model shows good predictive accuracy, we believe further improvements may be made through a better integration or design of the interaction between the core of the molecule and its substituents or the use of even more novel architectures.
Associated Content
Supporting Information
SuprCat_SI_v03_submission.pdf – database details, model architectures, model training details, extended fragment analysis, molecular coordinates.
Model code and analysis scripts can be found at https://github.com/rperezsoto/aS0T1_prediction_model.
Authort Information
Corresponding Author
Seonah Kim:
1301 Center Ave Mall, Chemistry B101, Campus Delivery
1872, Ft. Collins, CO 80523-1872
seonah.kim@colostate.edu
Robert Paton:
1301 Center Ave Mall, Chemistry B101, Campus Delivery
1872, Ft. Collins, CO 80523-1872
robert.paton@colostate.edu
Steven Lopez:
335 ISEC (Interdisciplinary Science and Engineering Complex), Boston, MA 02115
s.lopez@northeastern.edu
Author Contributions
Conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – original draft, review & editing: R.P.S., Conceptualization, methodology, formal analysis, investigation, writing – original draft, review & editing: M.V.P., Conceptualization, methodology, investigation, writing – original draft, review & editing: Sabari Kumar, methodology, investigation: L.A.G, methodology, investigation: C.L., methodology, writing – review & editing: E.S., conceptualization, formal analysis, writing review & editing: S.A.L., conceptualization, formal analysis, writing review & editing: R. S. P., conceptualization, funding acquisition, pro-ect administration, supervision, writing – review & editing: Seonah Kim.
Funding Sources
This research was supported by the National Science Foundation (NSF) Under the CCI Center for Sustainable Photoredox Catalysis (CHE-2318141).
Notes
The authors declare no competing interest
ACKNOWLEDGMENT
Seonah Kim acknowledges the the Colorado State University startup funds for PI (Seonah Kim) and the National Science Foundation under Grant No. CHE-2304658 for computing resources. Computer time was also provided by NSF ACCESS Grant No. CHE210028p (Seonah Kim) and Grant No. CHE180035p (Robert Paton).
REFERENCES
Strieth-Kalthoff, F.; James, M. J.; Teders, M.; Pitzer, L.; Glorius, F. Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47 (19), 7190–7202. https://doi.org/10.1039/C8CS00054A.
Melchiorre, P. Introduction: Photochemical Catalytic Processes. Chem. Rev. 2022, 122 (2), 1483–1484.
https://doi.org/10.1021/acs.chemrev.1c00993.
Introduction: Photochemistry in Organic Synthesis. Chem.Rev. 2016, 116 (17), 9629–9630.
https://doi.org/10.1021/acs.chemrev.6b00378.
Cabanero, D. C.; Rovis, T. Low-Energy Photoredox Catalysis.
Nat.Rev.Chem.2025, 9(1), 28–45. https://doi.org/10.1038/s41570-
024-00663-6.
Romero, N. A.; Nicewicz, D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116 (17), 10075–10166.
https://doi.org/10.1021/acs.chemrev.6b00057.
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113 (7), 5322–5363. https://doi.org/10.1021/cr300503r.
Popescu, M. V.; Paton, R. S. Dynamic Vertical Triplet Energies: Understanding and Predicting Triplet Energy Transfer. Chem2024, 10 (11), 3428–3443.
https://doi.org/10.1016/j.chempr.2024.07.001.
Park, W.; Komarov, K.; Lee, S.; Choi, C. H. Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory: Multireference
Advantages with the Practicality of Linear Response Theory. J. Phys.Chem. Lett. 2023, 14 (39), 8896–8908.
https://doi.org/10.1021/acs.jpclett.3c02296.
Nelson, T. R.; White, A. J.; Bjorgaard, J. A.; Sifain, A. E.; Zhang, Y.; Nebgen, B.; Fernandez-Alberti, S.; Mozyrsky, D.; Roitberg,
A. E.; Tretiak, S. Non-Adiabatic Excited-State Molecular Dynamics: Theory and Applications for Modeling Photophysics in Extended Molecular Materials. Chem. Rev. 2020, 120 (4), 2215–2287. https://doi.org/10.1021/acs.chemrev.9b00447.
Crespo-Otero, R.; Barbatti, M. Recent Advances and Perspectives on Nonadiabatic Mixed Quantum–Classical Dynamics. Chem. Rev. 2018, 118 (15), 7026–7068.
https://doi.org/10.1021/acs.chemrev.7b00577.
S. Paton, R. Real-Time Prediction of 1 H and 13 C Chemical Shifts with DFT Accuracy Using a 3D Graph Neural Network. Chem. Sci. 2021, 12(36), 12012–12026. https://doi.org/10.1039/D1SC03343C.
- Stubbs, C. D.; Kim, Y.; Quinn, E. C.; Pérez-Soto, R.; Chen, E. Y.-X.; Kim, S. Predicting Homopolymer and Copolymer Solubility through Machine Learning. Digit. Discov. 2024. https://doi.org/10.1039/D4DD00290C.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S. A. A.; Ballard, A. J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A. W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature2021, 596(7873),
583–589. https://doi.org/10.1038/s41586-021-03819-2.
- Kim, Y.; Cho, J.; Naser, N.; Kumar, S.; Jeong, K.; McCormick,
R. L.; St. John, P. C.; Kim, S. Physics-Informed Graph Neural Networks for Predicting Cetane Number with Systematic Data Quality Analysis. Proc. Combust. Inst. 2023, 39 (4), 4969–4978. https://doi.org/10.1016/j.proci.2022.09.059.
- Aldossary, A.; Campos-Gonzalez-Angulo, J. A.; Pablo-García, S.; Leong, S. X.; Rajaonson, E. M.; Thiede, L.; Tom, G.; Wang, A.; Avagliano, D.; Aspuru-Guzik, A. In Silico Chemical Experiments in the Age of AI: From Quantum Chemistry to Machine Learning and Back. Adv.Ma-ter. 2024, 36 (30), 2402369. https://doi.org/10.1002/adma.202402369.
- Sorkun, M. C.; Khetan, A.; Er, S. AqSolDB, a Curated Reference Set of Aqueous Solubility and 2D Descriptors for a Diverse Set of Compounds. Sci. Data 2019, 6 (1), 143. https://doi.org/10.1038/s41597-019-0151-1.
- Kim, Y.; Jung, H.; Kumar, S.; Paton, R. S.; Kim, S. Designing Solvent Systems Using Self-Evolving Solubility Databases and Graph Neural Networks. Chem. Sci. 2024, 15 (3), 923–939. https://doi.org/10.1039/D3SC03468B.
- Gelžinytė, E.; Öeren, M.; Segall, M. D.; Csányi, G. Transferable Machine Learning Interatomic Potential for Bond Dissociation Energy Prediction of Drug-like Molecule. ChemRxiv June 29, 2023. https://doi.org/10.26434/chemrxiv-2023-l85nf.
- St. John, P. C.; Guan, Y.; Kim, Y.; Kim, S.; Paton, R. S. Prediction of Organic Homolytic Bond Dissociation Enthalpies at near Chemical Accuracy with Sub-Second Computational Cost. Nat. Commun . 2020, 11 (1), 2328. https://doi.org/10.1038/s41467-020-16201-z.
- V, S. S. S.; Kim, Y.; Kim, S.; John, P. C. S.; Paton, R. S. Expan-
sion of Bond Dissociation Prediction with Machine Learning to Medicinally and Environmentally Relevant Chemical Space. Digit. Discov.2023. https://doi.org/10.1039/D3DD00169E.
- Casetti, N.; Alfonso-Ramos, J. E.; Coley, C. W.; Stuyver, T. Combining Molecular Quantum Mechanical Modeling and Machine Learning for Accelerated Reaction Screening and Discovery. Chem. –Eur. J. 2023, 29 (60), e202301957.
https://doi.org/10.1002/chem.202301957.
- Lunger, J. R.; Karaguesian, J.; Chun, H.; Peng, J.; Tseo, Y.; Shan, C. H.; Han, B.; Shao-Horn, Y.; Gómez-Bombarelli, R. Towards Atom-Level Understanding of Metal Oxide Catalysts for the Oxygen Evolution Reaction with Machine Learning. NpjComput.Mater.2024, 10 (1), 1–11. https://doi.org/10.1038/s41524-024-01273-y.
- Ferri, P.; Li, C.; Schwalbe-Koda, D.; Xie, M.; Moliner, M.; Gómez-Bombarelli, R.; Boronat, M.; Corma, A. Approaching Enzymatic Catalysis with Zeolites or How to Select One Reaction Mechanism Competing with Others. Nat. Commun. 2023, 14 (1), 2878. https://doi.org/10.1038/s41467-023-38544-z.
- Abreha, B. G.; Agarwal, S.; Foster, I.; Blaiszik, B.; Lopez, S. A. Virtual Excited State Reference for the Discovery of Electronic Materials Database: An Open-Access Resource for Ground and Excited State Properties of Organic Molecules. J. Phys. Chem. Lett. 2019, 10 (21), 6835–6841. https://doi.org/10.1021/acs.jpclett.9b02577.
- Schlosser, L.; Rana, D.; Pflüger, P.; Katzenburg, F.; Glorius, F. EnTdecker − A Machine Learning-Based Platform for Guiding Substrate Discovery in Energy Transfer Catalysis. J. Am. Chem. Soc.2024. https://doi.org/10.1021/jacs.4c01352.
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16 Rev. C.01, 2016.
- Ertl, P. An Algorithm to Identify Functional Groups in Organic Molecules. J. Cheminformatics 2017, 9 (1), 36. https://doi.org/10.1186/s13321-017-0225-z.
- Yadav, L. D. S. Ultraviolet (UV) and Visible Spectroscopy. In OrganicSpectroscopy; Yadav, L. D. S., Ed.; Springer Netherlands: Dordrecht, 2005; pp 7–51. https://doi.org/10.1007/978-1-4020-2575- 4_2.
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615–6620.
https://doi.org/10.1039/B810189B.
- Scalmani, G.; Frisch, M. J. Continuous Surface Charge Polar- izable Continuum Models of Solvation. I. General Formalism. J.Chem.Phys. 2010, 132 (11), 114110. https://doi.org/10.1063/1.3359469.
- Becke, A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. https://doi.org/10.1063/1.464913.
- Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Sal- vetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37 (2), 785–789.
https://doi.org/10.1103/PhysRevB.37.785.
- Vosko, S. H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calcula- tions: A Critical Analysis. Can. J. Phys. 1980, 58 (8), 1200–1211. https://doi.org/10.1139/p80-159.
- Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism
Spectra Using Density Functional Force Fields. J.Phys.Chem.1994, 98
(45), 11623–11627. https://doi.org/10.1021/j100096a001.
- Neese, F. The ORCA Program System. WIREs Comput. Mol.Sci. 2012, 2 (1), 73–78. https://doi.org/10.1002/wcms.81.
- Tingle, B. I.; Tang, K. G.; Castanon, M.; Gutierrez, J. J.; Khurelbaatar, M.; Dandarchuluun, C.; Moroz, Y. S.; Irwin, J. J. ZINC- 22─A Free Multi-Billion-Scale Database of Tangible Compounds for Ligand Discovery. J. Chem. Inf. Model. 2023, 63 (4), 1166–1176. https://doi.org/10.1021/acs.jcim.2c01253.
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55(1), 117–129. https://doi.org/10.1016/0301-0104(81)85090-2.
- Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, PV. R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calcu- lations. III. The 3-21+G Basis Set for First-Row Elements, Li–F. J. Com-put. Chem. 1983, 4 (3), 294–301.
https://doi.org/10.1002/jcc.540040303.
- Hariharan, P. C.; Pople, J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor.Chim.Acta 1973, 28 (3), 213–222. https://doi.org/10.1007/BF00533485.
- Zhao, Y.; Truhlar, D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Non- covalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Function- als and 12 Other Functionals. Theor. Chem. Acc. 2008, 120 (1), 215–241. https://doi.org/10.1007/s00214-007-0310-x.
- Hehre, W. J.; Ditchfield, R.; Pople, J. A. Self—Consistent Mo- lecular Orbital Methods. XII. Further Extensions of Gaussian—Type Ba- sis Sets for Use in Molecular Orbital Studies of Organic Molecules. J.Chem. Phys. 1972, 56 (5), 2257–2261.
https://doi.org/10.1063/1.1677527.
- Dunning, T. H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydro- gen. J. Chem. Phys. 1989, 90 (2), 1007–1023.
https://doi.org/10.1063/1.456153.
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D. G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An Improved Linear Scaling Per- turbative Triples Correction for the Domain Based Local Pair-Natural Orbital Based Singles and Doubles Coupled Cluster Method [DLPNO- CCSD(T)]. J. Chem. Phys. 2018, 148 (1), 011101.
https://doi.org/10.1063/1.5011798.
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E. F.; Neese, F. A New Near-Linear Scaling, Efficient and Accurate, Open-Shell Do- main-Based Local Pair Natural Orbital Coupled Cluster Singles and Doubles Theory. J. Chem. Phys. 2017, 146 (16), 164105. https://doi.org/10.1063/1.4981521.
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E. F.; Neese, F. Sparse Maps—A Systematic Infrastructure for Reduced-Scaling Elec- tronic Structure Methods. II. Linear Scaling Domain Based Pair Natural Orbital Coupled Cluster Theory. J.Chem.Phys.2016, 144(2), 024109. https://doi.org/10.1063/1.4939030.
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Nat- ural Orbitals. J. Chem. Phys. 2013, 139 (13), 134101. https://doi.org/10.1063/1.4821834.
- Riplinger, C.; Neese, F. An Efficient and near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster Method. J. Chem.Phys. 2013, 138 (3), 034106. https://doi.org/10.1063/1.4773581.
- Ramakrishnan, R.; Dral, P. O.; Rupp, M.; Lilienfeld, O. A. von. Big Data Meets Quantum Chemistry Approximations: The $Δ$-Ma- chine Learning Approach. J.Chem.TheoryComput.2015, 11(5), 20872096. https://doi.org/10.1021/acs.jctc.5b00099.
- Chen, X.; Li, P.; Hruska, E.; Liu, F. Δ-Machine Learning for Quantum Chemistry Prediction of Solution-Phase Molecular Proper- ties at the Ground and Excited States. Phys.Chem.Chem.Phys.2023, 25 (19), 13417–13428. https://doi.org/10.1039/D3CP00506B.
- Yang, C.; Zhang, Y. Delta Machine Learning to Improve Scor- ing-Ranking-Screening Performances of Protein–Ligand Scoring Func- tions. J. Chem. Inf. Model. 2022, 62 (11), 2696–2712. https://doi.org/10.1021/acs.jcim.2c00485.
- Grumet, M.; von Scarpatetti, C.; Bučko, T.; Egger, D. A. Delta Machine Learning for Predicting Dielectric Properties and Raman Spectra. J. Phys. Chem. C 2024, 128 (15), 6464–6470.